Whole Human Genome Sequencing and Analysis Report

Personal Genome Annotation System
This is a summary report for NA####
The products from Diagnomics do not provide any medical advice, diagnosis or treatment. Analyses and reports provided by Diagnomics are for informational purposes only and are subject to change. You should consult your physician if you have questions regarding any medical condition. The results and analysis presented have not been cleared or approved by the FDA or similar government institutions.
Physician and Patient Information
Physician name: #### ####, M.D.
Address: #### ####
#### ####
Patient name: #### ####
Patient DOB: #### ####
Patient gender: F
Patient ethnicity: Caucasian
Summary of the Report
Diagnomics’ Personal Genome Annotation System (PAS) is a comprehensive pipeline for personal genome annotation available for genome sequencing to date from Targeted, Exom, and Whole Genome Sequencing platforms. The PAS system provides highly customizable genome variation report combined with various package of genetic annotation including gene, transcripts and amino acid variation, functional variation analysis, rare genetic disease, common disease risk analysis as well as drug response based on individual genetic make-up. The information provide valuable insights to researchers, individuals and physicians who are interested in exploring personal genome information for personalized medicine.
Inherited Genetic Conditions
A genetic disorder is an illness caused by one or more abnormalities in the genome, especially a condition that is present from birth (congenital). Genetic disorders are heritable, and are passed down from the parents' genes. Many of rare genetic disorder are the result of a single mutated gene. Over 4000 human diseases are caused by single gene defects. Some of rare genetic disorder is affected by dominant condition that only one mutated copy of the gene is necessary for person to be affected. Many genetic disorders are recessive condition that two copies of the gene must be mutated to be affected. The unaffected parents who have one copy of the mutated gene (Carrier) have a 25% change with each pregnancy of having a child affected by the disorder.
Some Genetic disorders may also be complex, multifactorial or polygenic, meaning they are likely associated with the effects of multiple genes in combination with lifestyles and environmental factors. Although complex disorders often cluster in families, they do not have a clear-cut pattern of inheritance.
Genetic Disease and Carrier Screening
The genetic disease testing is used to confirm a particular condition is suspected based on physical mutations and symptoms. The results of the test can influence a person's choices about health care and the management of the disease. Carrier testing is used to identify people who carry one copy of a gene mutation that, when present in two copies, causes a genetic disorder. This type of testing is offered to individuals who have a family history of a genetic disorder and to people in ethnic groups with an increased risk of specific genetic conditions. If both parents are tested, the test can provide information about a couple's risk of having a child with a genetic condition.
| #CHROM | POS | ID | REF | ALT | QUAL | FILTER | CODING_CONSEQUENCE | AA_CHANGE | DNA_CHANGE | GENE_HGNC | SIFT | POLYPHEN | COSMICID | GENE_OMIM | GENE_TITLE_OMIM | MIM_OMIM | DISORDERS_OMIM | AUTOSOMAL_DOMINANT_CLINVAR | SIGNIFICANCE_CLINVAR | CLNDBN_CLINVAR | CLNDSDBID_CLINVAR | CLNACC_CLINVAR | CLNACC_CLINVAR_LINKS | |||||
|
4 |
C |
G |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:23,26:49:99:99:60:700,0,745:0.531 |
missense_variant |
L/V |
Ctt/Gtt |
tolerated(1) |
benign(0.218) |
0 |
Cytochrome P450, family 4, subfamily V, poypeptide 2 |
Bietti crystalline corneoretinal dystrophy, 210370 (3) |
0 |
pathogenic |
Bietti crystalline corneoretinal dystrophy |
NBK91457:C1859486:210370 |
RCV000032544.2 |
|||||||||
|
22 |
G |
C |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,44:44:99:99:60:1633,129,0:1.000 |
missense_variant |
T/S |
aCt/aGt |
tolerated(0.41) |
benign(0) |
0 |
0 |
pathogenic |
Metachromatic leukodystrophy |
NBK1130:C0023522:250100:512:396338004 |
RCV000020311.1 |
|||||||||||
|
1 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:36,22:58:99:99:60:727,0,1230:0.379 |
stop_gained&splice_region_variant |
Q/* |
Caa/Taa |
AMPD1 |
0 |
0 |
pathogenic |
Muscle AMP deaminase deficiency |
C0268123:9105005 |
RCV000019933.1 |
||||||||||||
|
1 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:36,22:58:99:99:60:727,0,1230:0.379 |
stop_gained&splice_region_variant |
Q/* |
Caa/Taa |
YES |
AMPD1 |
0 |
0 |
pathogenic |
Muscle AMP deaminase deficiency |
C0268123:9105005 |
RCV000019933.1 |
|||||||||||
|
1 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:36,22:58:99:99:60:727,0,1230:0.379 |
stop_gained&splice_region_variant |
Q/* |
Caa/Taa |
0 |
0 |
pathogenic |
Muscle AMP deaminase deficiency |
C0268123:9105005 |
RCV000019933.1 |
|||||||||||||
|
11 |
C |
T |
99 |
TruthSensitivityTranche99.00to99.90 |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:19,14:33:99:99:46:226,0,251:0.424 |
missense_variant |
A/V |
gCc/gTc |
tolerated(0.71) |
benign(0.049) |
1 |
GENE=SAA1;STRAND=+;CDS=c.209C>T;AA=p.A70V;CNT=1 |
0 |
pathogenic |
Serum amyloid a variant |
. |
RCV000019736.1 |
||||||||||
|
12 |
C |
T |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:18,17:35:99:99:60:566,0,611:0.486 |
missense_variant |
D/N |
Gac/Aac |
tolerated(1) |
benign(0.002) |
1 |
GENE=ART4;STRAND=-;CDS=c.793G>A;AA=p.D265N;CNT=1 |
ADP-ribosyltransferase-4 (Dombrock blood group) |
[Blood group, Dombrock] (3) |
0 |
pathogenic |
DOMBROCK BLOOD GROUP |
. |
RCV000019304.1 |
||||||||
|
22 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:21,27:48:99:99:58:800,0,561:0.563 |
missense_variant |
P/S |
Cca/Tca |
deleterious(0.03) |
possibly_damaging(0.581) |
0 |
Cytochrome P450, subfamily IID, polypeptide 6 |
{Debrisoquine sensitivity}, 608902 (3); {Codeine sensitivity}, 608902 (3) |
1 |
pathogenic |
Debrisoquine\x2c poor metabolism of |
C1837156 |
RCV000018389.1 |
|||||||||
|
5 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:13,10:23:99:99:60:308,0,441:0.435 |
missense_variant |
G/R |
Ggg/Agg |
tolerated(0.35) |
possibly_damaging(0.515) |
0 |
0 |
pathogenic |
Cancer progression and tumor cell motility |
. |
RCV000017723.1 |
|||||||||||
|
5 |
G |
A |
78 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,27:27:78.23:78:60:1011,78,0:1.000 |
missense_variant&NMD_transcript_variant |
V/I |
Gtc/Atc |
tolerated(0.73) |
benign(0) |
1 |
GENE=IL7R;STRAND=+;CDS=c.412G>A;AA=p.V138I;CNT=1 |
Interleukin-7 receptor |
Severe combined immunodeficiency, T-cell negative, B-cell/natural killer cell-positive type, 608971 (3) |
0 |
pathogenic |
Severe combined immunodeficiency\x2c autosomal recessive\x2c T cell-negative\x2c B cell-positive\x2c NK cell-positive |
C1837028:608971 |
RCV000015965.1 |
||||||||
|
5 |
T |
C |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,39:39:99:99:60:1452,111,0:1.000 |
missense_variant&NMD_transcript_variant |
I/T |
aTc/aCc |
tolerated(0.7) |
benign(0) |
1 |
GENE=IL7R;STRAND=+;CDS=c.197T>C;AA=p.I66T;CNT=1 |
Interleukin-7 receptor |
Severe combined immunodeficiency, T-cell negative, B-cell/natural killer cell-positive type, 608971 (3) |
0 |
pathogenic |
Severe combined immunodeficiency\x2c autosomal recessive\x2c T cell-negative\x2c B cell-positive\x2c NK cell-positive |
C1837028:608971 |
RCV000015964.1 |
||||||||
|
1 |
C |
T |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:8,21:29:99:99:60:667,0,186:0.724 |
missense_variant |
V/I |
Gta/Ata |
tolerated(0.72) |
unknown(0) |
0 |
Syndecan 3 |
{Obesity, association with}, 601665 (3) |
0 |
pathogenic |
Obesity\x2c association with |
. |
RCV000013593.1 |
|||||||||
|
7 |
T |
C |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:18,8:26:99:99:60:186,0,573:0.308 |
missense_variant |
I/V |
Atc/Gtc |
tolerated(0.07) |
possibly_damaging(0.84) |
0 |
Taste receptor, type 2, member 38 |
[Phenylthiocarbamide tasting], 171200 (3) |
0 |
pathogenic |
Phenylthiocarbamide tasting |
C1868398 |
RCV000003040.1 |
|||||||||
|
17 |
A |
C |
90 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,35:36:90.24:90:60:1108,90,0:1.000 |
missense_variant |
I/S |
aTc/aGc |
tolerated(0.21) |
benign(0) |
0 |
Integrin, alpha-2b (platelet glycoprotein IIb of IIb/IIIa complex, antigen CD41B) |
Glanzmann thrombasthenia, 273800 (3); Thrombocytopenia, neonatal alloimmune, BAK antigen related (3); Bleeding disorder, platelet-type, 16, autosomal dominant, 187800 (3) |
1 |
pathogenic |
Bak platelet-specific antigen |
. |
RCV000003025.1 |
|||||||||
|
11 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:20,18:39:99:99:60:587,0,583:0.474 |
missense_variant |
E/K |
Gag/Aag |
tolerated(1) |
benign(0) |
0 |
0 |
pathogenic |
Dopamine receptor d2\x2c reduced brain density of |
C1837439 |
RCV000002186.1 |
|||||||||||
|
10 |
A |
C |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:27,21:48:99:99:60:659,0,923:0.438 |
missense_variant |
E/D |
gaA/gaC |
tolerated(0.84) |
benign(0) |
0 |
Storkhead box 1 |
Preeclampsia/eclampsia 4, 609404 (3) |
0 |
pathogenic |
Preeclampsia/eclampsia 4 |
C1836255:609404 |
RCV000001790.1 |
|||||||||
|
4 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:22,18:40:99:99:59:523,0,744:0.450 |
missense_variant |
R/H |
cGc/cAc |
tolerated(0.15) |
benign(0.008) |
0 |
B-cell scaffold protein with ankyrin repeats 1 |
{Systemic lupus erythematosus, association with}, 152700 (3) |
0 |
pathogenic |
Systemic lupus erythmatosus\x2c association with |
. |
RCV000001331.1 |
|||||||||
|
11 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:24,20:44:99:99:60:641,0,750:0.455 |
missense_variant |
G/E |
gGg/gAg |
tolerated(0.46) |
benign(0.01) |
0 |
Two-pore segment channel 2 |
[Skin/hair/eye pigmentation 10, blond/brown hair], 612267 (3) |
0 |
pathogenic |
Skin/hair/eye pigmentation\x2c variation in\x2c 10 |
C2677088:612267 |
RCV000000764.1 |
|||||||||
|
11 |
A |
T |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:18,18:36:99:99:60:537,0,595:0.500 |
missense_variant |
M/L |
Atg/Ttg |
tolerated(1) |
benign(0.01) |
0 |
Two-pore segment channel 2 |
[Skin/hair/eye pigmentation 10, blond/brown hair], 612267 (3) |
0 |
pathogenic |
Skin/hair/eye pigmentation\x2c variation in\x2c 10 |
C2677088:612267 |
RCV000000763.1 |
|||||||||
|
12 |
T |
C |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:26,16:42:99:99:60:496,0,836:0.381 |
missense_variant |
Y/C |
tAc/tGc |
deleterious(0) |
probably_damaging(1) |
0 |
Phenylalanine hydroxylase |
Phenylketonuria, 261600 (3); [Hyperphenylalaninemia, non-PKU mild], 261600 (3) |
0 |
pathogenic |
Hyperphenylalaninemia\x2c non-pku |
C0751435 |
RCV000000624.1 |
|||||||||
|
12 |
A |
G |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,41:41:99:99:60:1501,120,0:1.000 |
missense_variant |
S/G |
Agc/Ggc |
tolerated(0.58) |
benign(0) |
0 |
HNF1 homeobox B |
MODY, type III, 600496 (3); {Diabetes mellitus, noninsulin-dependent, 2}, 125853 (3); {Diabetes mellitus, insulin-dependent}, 222100 (3); Hepatic adenoma, somatic, 142330 (3); Renal cell carcinoma, 144700 (3); Diabetes mellitus, insulin-dependent, 20, 612520 (3) |
1 |
pathogenic,untested |
Maturity-onset diabetes of the young\x2c type 3,. |
C1838100:600496:552,. |
RCV000016077.1,. |
|||||||||
|
5 |
C |
G |
84 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,29:29:84.27:84:60:1096,84,0:1.000 |
missense_variant |
L/F |
ttG/ttC |
deleterious(0) |
probably_damaging(0.996) |
0 |
Solute carrier family 45, member 2 |
Oculocutaneous albinism, type IV, 606574 (3); [Skin/hair/eye pigmentation 5, black/nonblack hair], 227240 (3); [Skin/hair/eye pigmentation 5, dark/fair skin], 227240 (3); [Skin/hair/eye pigmentation 5, dark/light eyes], 227240 (3) |
0 |
pathogenic,untested |
Skin/hair/eye pigmentation\x2c variation in\x2c 5,. |
C2673584:227240,. |
RCV000004763.1,. |
|||||||||
|
3 |
C |
T |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:26,19:45:99:99:57:635,0,687:0.422 |
missense_variant |
A/T |
Gca/Aca |
tolerated(0.48) |
benign(0.022) |
0 |
Butyrylcholinesterase |
Apnea, postanesthetic (3) |
0 |
pathogenic|non-pathogenic|pathogenic|pathogenic |
Bche\x2c k variant|BCHE\x2c QUANTITATIVE K POLYMORPHISM|CHE*539T|BCHE*539T |
.|.|.|. |
RCV000014120.1|RCV000014121.1|RCV000014122.1|RCV000014123.1 |
|||||||||
|
3 |
C |
T |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:19,16:35:99:99:60:532,0,568:0.457 |
missense_variant |
P/S |
Cct/Tct |
tolerated(0.16) |
benign(0.03) |
0 |
Transferrin |
Atransferrinemia, 209300 (3) |
0 |
pathogenic|other |
Transferrin variant c1/c2|Alzheimer disease\x2c susceptibility to |
.|. |
RCV000013451.1|RCV000013452.1 |
|||||||||
|
19 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,40:40:99:99:60:1396,114,0:1.000 |
missense_variant |
P/L |
cCg/cTg |
tolerated(0.77) |
unknown(0) |
0 |
Transforming growth factor, beta-1 |
Camurati-Engelmann disease, 131300 (3); {Cystic fibrosis lung disease, modifier of}, 219700 (3) |
0 |
pathogenic|other|non-pathogenic |
Cystic fibrosis|Breast cancer\x2c invasive\x2c susceptibility to|Diaphyseal dysplasia |
NBK1250:C0010674:219700:586:190905008|.|NBK1156:C0011989:131300:1328:34643004 |
RCV000013360.1|RCV000013361.1|RCV000032141.1 |
|||||||||
|
3 |
G |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:14,13:27:99:99:60:433,0,393:0.481 |
missense_variant |
T/M |
aCg/aTg |
deleterious(0) |
probably_damaging(1) |
0 |
X transporter protein 3 |
Hyperglycinuria, 138500 (3); Iminoglycinuria, digenic, 242600 (3) |
1 |
pathogenic|pathogenic |
Hyperglycinuria|Iminoglycinuria\x2c digenic |
C0543541:138500|. |
RCV000005117.1|RCV000005118.1 |
|||||||||
|
1 |
T |
C |
87 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
1/1:0,33:33:87.28:87:58:1163,87,0:1.000 |
missense_variant |
Q/R |
cAa/cGa |
tolerated(1) |
benign(0) |
0 |
Coagulation factor V (proaccelerin, labile factor) |
Factor V deficiency, 227400 (3); {Thrombophilia, susceptibility to, due to factor V Leiden}, 188055 (3); {Stroke, ischemic, susceptibility to}, 601367 (3); {Budd-Chiari syndrome}, 600880 (3); Thrombophilia due to activated protein C resistance, 188055 (3); {Pregnancy loss, recurrent, susceptibility to, 1}, 614389 (3) |
1 |
pathogenic|untested|other|other,untested |
Thrombophilia due to factor V leiden|Ischemic stroke\x2c susceptibility to|Budd-Chiari syndrome\x2c susceptibility to|Recurrent abortion,. |
C2674152|.|.|C0000809:614389:102878001,. |
RCV000000674.1|RCV000000675.1|RCV000000676.1|RCV000023935.1,. |
|||||||||
|
7 |
C |
A |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:18,15:33:99:99:60:466,0,524:0.455 |
missense_variant |
R/L |
cGc/cTc |
tolerated(0.22) |
possibly_damaging(0.786) |
0 |
Potassium voltage-gated channel, subfamily H, member 2 (human ether-a-go-go-related gene) |
Long QT syndrome-2, 613688 (3); {Long QT syndrome-2, acquired, susceptibility to}, 613688 (3); Short QT syndrome-1, 609620 (3) |
1 |
probable-non-pathogenic |
Cardiac arrhythmia |
C0003811:115000 |
RCV000030101.1 |
|||||||||
|
13 |
C |
G |
99 |
PASS |
GT:AD:DP:GQ:GQX:MQ:PL:VF |
0/1:29,24:53:99:99:60:801,0,890:0.453 |
missense_variant |
V/L |
Gtg/Ctg |
tolerated(0.36) |
benign(0.003) |
0 |
ATPase, Cu++ transporting, beta polypeptide |
Wilson disease, 277900 (3) |
0 |
probable-non-pathogenic |
Wilson's disease |
NBK1512:C0019202:277900:905:88518009 |
RCV000029351.1 |
|||||||||
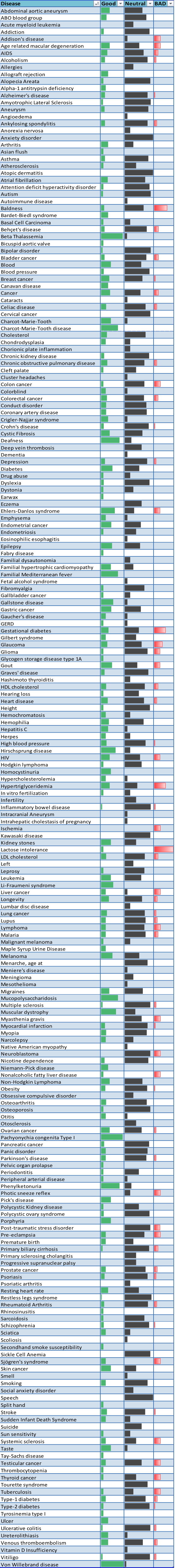
Predictive Disease Risk Analysis
These types of testing are used to detect gene mutations associated with disorders that appear after birth, often later in life. These tests can be helpful to people who have a family member with a genetic disorder, but who have no features of the disorder themselves at the time of testing. Predictive testing can identify mutations that increase a person's risk of developing disorders with a genetic basis, such as certain types of cancer. For example, an individual with a mutation in BRCA1 has a 65% cumulative risk of breast cancer. The results of predictive and presymptomatic testing can provide information about a person’s risk of developing a specific disorder and help with making decisions about medical care.
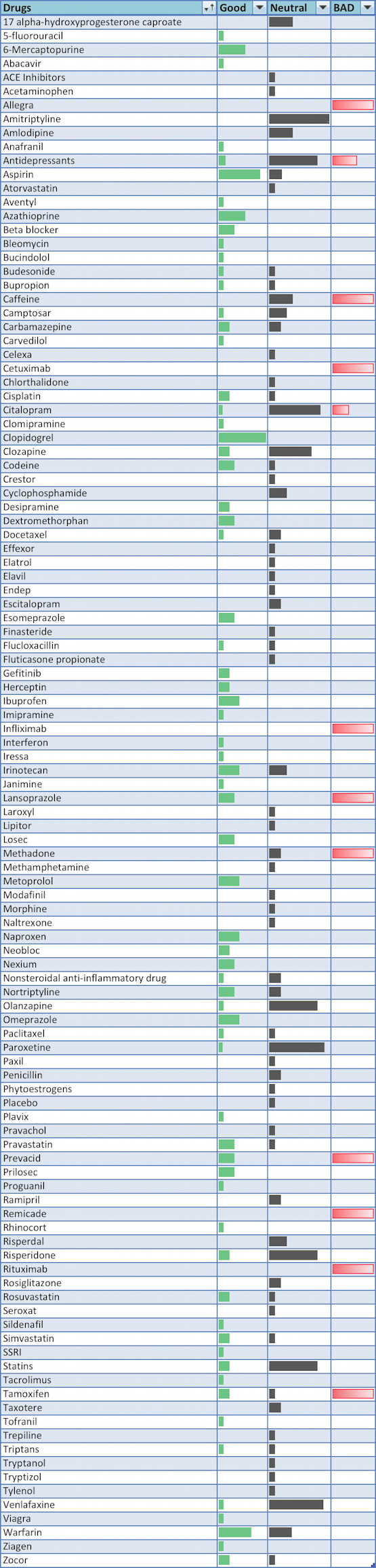
Adverse Drug Reaction And Efficacy Analysis
Variation Analysis
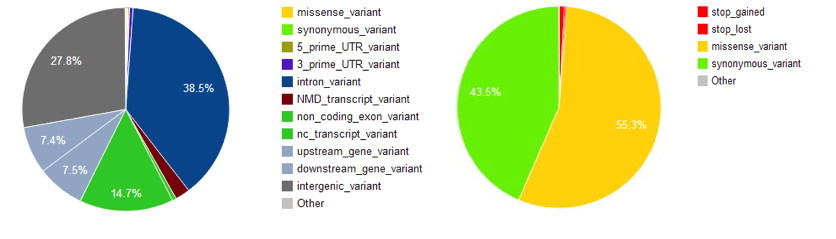
The interpretation of novel missense variants is a challenge with increasing numbers of such variants being identified and a responsibility to report the findings in the context of all available scientific evidence. Various in silico bioinformatic tools and database have been developed that helps the understanding of such variations in the context of pathogenicity of missense variations.
SIFT
SIFT (Sorting Intolerant From Tolerant) is a program that predicts whether an amino acid substitution affects protein function. SIFT prediction is based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences, collected through PSI-BLAST. SIFT can be applied to predict naturally occurring non-synonymous polymorphisms. SIFT has been applied to human variant databases and was able to distinguish mutations involved in disease from neutral polymorphisms.
| SIFT Prediction Count | Variations | Percentage |
| Deleterious | 3,891 | 23.8% |
| Tolerated | 12,437 | 76.2% |
| Total Number | 16,328 | 100.0% |
PolyPhen
PolyPhen (Polymorphism Phenotyping) is a software tool which predicts possible impact of amino acid substitutions on the structure and function of human proteins using straightforward physical and evolutionary comparative considerations.
| PolyPhen Prediction Count | Variations | Percentage |
| Probably damaging | 1,903 | 10.2% |
| Possibly damaging | 1,926 | 10.3% |
| Unknown | 3,274 | 17.5% |
| Benign | 11,616 | 62.1% |
| Total Number | 18,719 | 100.0% |
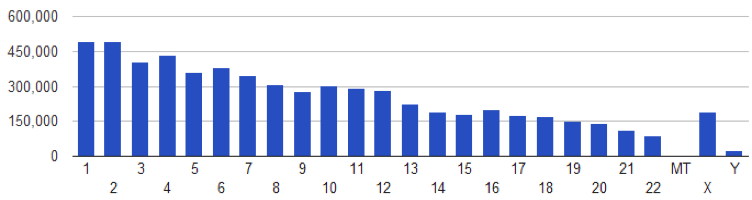
Variations by Chromosome
Sample and Sequencing Information
Sample barcode: #### ####, M.D.
Sample type: DNA
Sample origine: Germline
Collection date: #### ####
Receiving date: #### ####
Report date: #### ####
Reference Genome: NCBI37
SNP Assessment
| Total | Het/Hom | % in dbSNP | % in Genes | % in Coding |
| 3,325,664 | 1.40 | 97.74% | 45.09% | 0.62% |
Variant Statistics
| SNVs | |
| Total Number | 3,325,664 |
| Number in Genes | 1,499,669 |
| Number in Coding Regions | 20,697 |
| Number in UTRs | 24,709 |
| Splice Site Region | 2,951 |
| Stop Gained | 74 |
| Stop Lost | 16 |
| Non-synonymous | 9,749 |
| Synonymous | 10,858 |
| Mature miRNA | 41 |
The SNP assessment and statistics tables above provide a summary of variant loci, overlap with dbSNP, and consequences of variants.
Sequencing Library and Read Specifications
Below are statistics that describe the short-read sequencing library prepared from the submitted sample. Also indicated are sequencing read type and read length.
Fragment Length Median: 334Fragment Length SD: 62
Read Type: Paired
Read 1 Length: 100
Read 2 Length: 100
Data Volume and Quality
| Yield (Gigabases) | % Bases ≥ Q30 | % Bases Aligned | |
| Passing Filter | 117.39 | 87.10% | 88.00% |
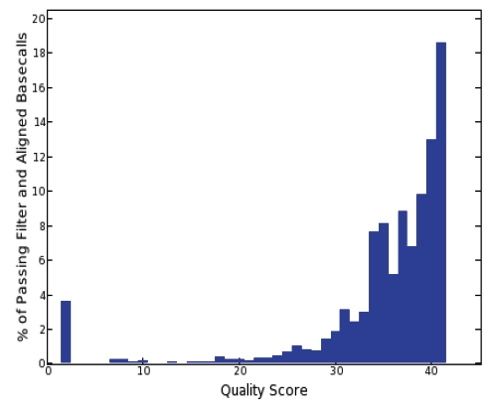
The table above provides a summary of the sequencing experiment showing the total volume of bases sequenced, the fraction of bases with Phred-scale qualities greater than or equal to 30 and the fraction of bases that aligned to the reference human genome. Figure below shows the distribution of aligned basecall qualities.
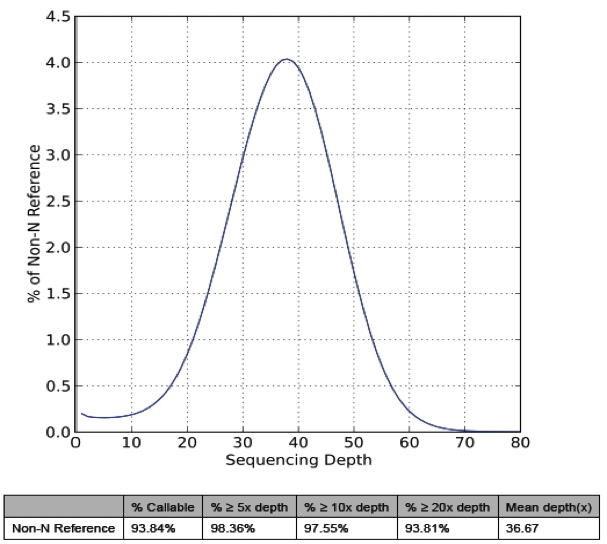
Non-N Reference Coverage Distribution
The figure and table below summarize coverage of ungapped regions of the human reference genome with unique DNA fragments. Each base in the reference genome is sequenced and mapped to the human genome an average of 30 times (x), represented by the mean depth. Also shown are the fraction of bases that are covered at or above 5x and 10x, and the fraction of bases where a genotype call was made.
Sequencing Methodology
Sequence was generated from extracted DNA. DNA extracted was sequenced using the Illumina HiSeq 2500 sequencer. Briefly, DNA was fragmented, and the fragments were attached to the surface of a glass microscope slide. The fragments were then sequenced using fluorescently labeled nucleotides, which were excited by a laser and imaged using digital equipment.
These fragments were then assessed for quality using a variety of metrics to ensure that only robust sequences were analyzed. Fragments were aligned to the NCBI reference sequence. Fragments that aligned to more than one region of the reference genome were excluded from the report. Additionally, fragments were excluded from the analysis on the basis of quality and alignment scores. Each nucleotide site reported was sequenced an average of 30 times, so there was on average 30-fold redundancy for each base pair reported for normal sample and average 90-fold redundancy for tumor sample. Additionally, no positions were called when the genotype quality score was less than 30 or depth was less than 6. Other types of variants are not called or validated in the Clinical Services Laboratory.
We made rigorous efforts to report a high quality consensus sequence, however due to a variety of factors, this genome report should not be considered complete or perfect. The regions of the genome not reported here include regions where the human reference genome has not been completely resolved, or are duplications of genetic regions make it impossible to align the fragments accurately. Although the error rates for this kind of technology are believed to be quite low, the sequence provided here cannot be considered as diagnostic.
Clinical action for any variants of potential medical concern should only be considered after further investigation confirms the presence of the variant using alternative, more focused testing specifically developed for that variant.
This test was developed and its performance characteristics determined by Clinical Services Laboratory. It has not been cleared or approved by the U.S. Food and Drug Administration.
References
- Bentley et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53-59.
- Wheeler et al. (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452:872-876.
- Levy et al. (2007) The diploid genome sequence of an individual human. PLoS Biology 5(10):e254.
- Wang et al. (2008) The diploid genome sequence of an Asian individual. Nature 456:60-66.
- The International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931-945.